在本教程中,我们将学习对不同条件刺激和处理的样品进行整合比较分析,用于探究在不同条件刺激和处理下样本之间的响应和差异。
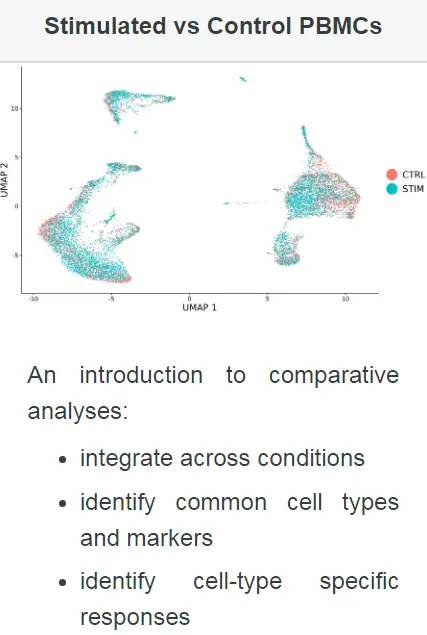
本次示例中,我们使用了Kang等人(2017)的两组PBMC的单细胞测序数据。在该实验中,将PBMC分为刺激组和对照组,并用干扰素β治疗刺激组。对干扰素的反应引起细胞类型特异性基因表达的变化,这使得对所有数据进行联合分析变得困难,并且细胞会根据刺激条件和细胞类型的不同而进行聚类分群。在这里,我们证明了我们的整合策略,如Stuart和Butler等人(2018年)所述,用于对不同条件刺激的样品进行整合分析,以促进常见细胞类型的鉴定并进行比较分析。
整合比较分析的目标:
- 鉴定在不同条件刺激下两个数据集中都存在的细胞类型
- 获得在对照和刺激组细胞中均保守的细胞类型标记基因
- 比较两个数据集,以找到对刺激响应的特定细胞类型
加载所需的R包和数据集
library(Seurat)library(SeuratData)library(cowplot)# 使用SeuratData包安装InstallData("ifnb")
构建Seurat对象
# 加载数据集data("ifnb")# 根据不同的处理条件分割对象ifnb.list <- SplitObject(ifnb, split.by = "stim")# 分别对两个样本进行数据归一化ifnb.list <- lapply(X = ifnb.list, FUN = function(x) {x <- NormalizeData(x)x <- FindVariableFeatures(x, selection.method = "vst", nfeatures = 2000)})
进行数据整合分析
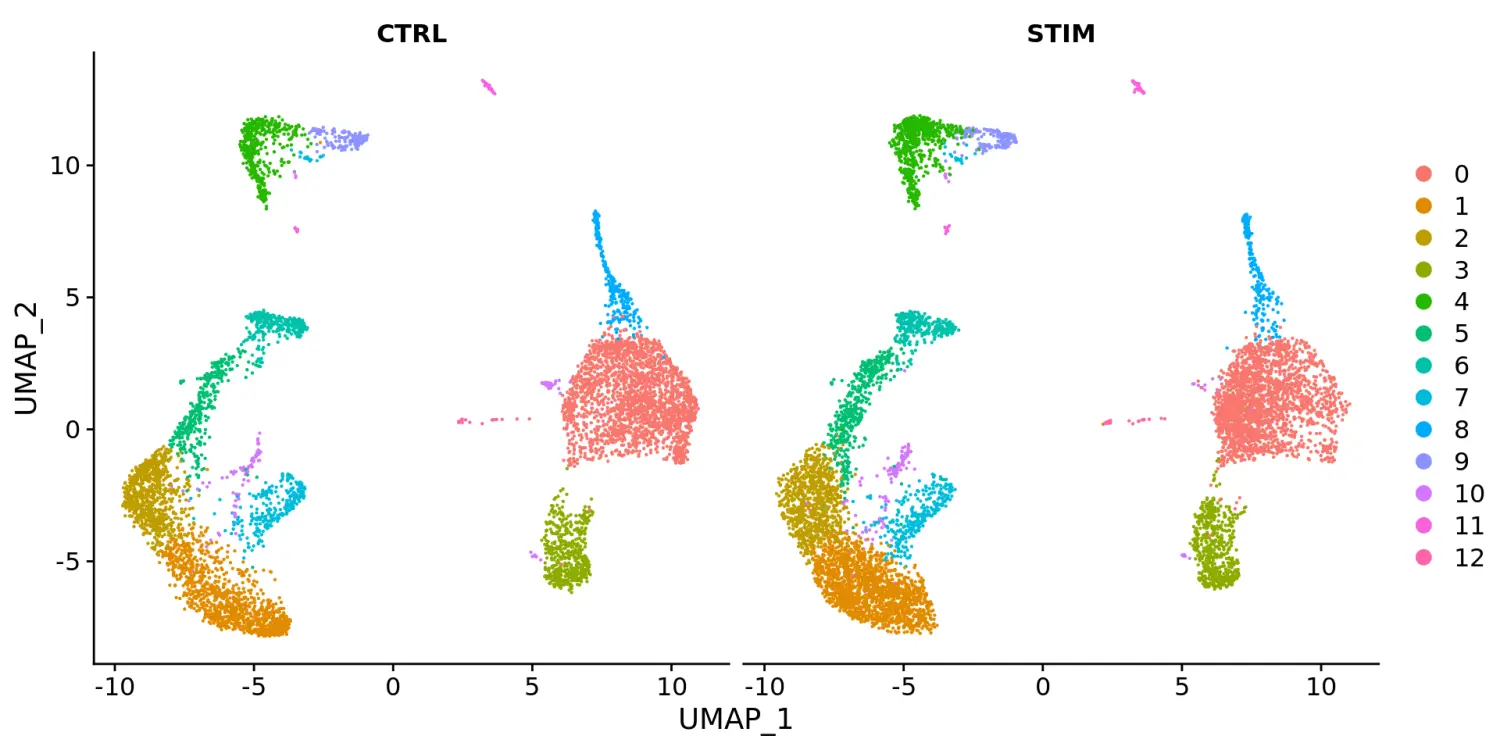
# 识别整合的anchorsimmune.anchors <- FindIntegrationAnchors(object.list = ifnb.list, dims = 1:20)# 使用IntegrateData函数进行整合immune.combined <- IntegrateData(anchorset = immune.anchors, dims = 1:20)DefaultAssay(immune.combined) <- "integrated"# Run the standard workflow for visualization and clusteringimmune.combined <- ScaleData(immune.combined, verbose = FALSE)immune.combined <- RunPCA(immune.combined, npcs = 30, verbose = FALSE)# t-SNE and Clusteringimmune.combined <- RunUMAP(immune.combined, reduction = "pca", dims = 1:20)immune.combined <- FindNeighbors(immune.combined, reduction = "pca", dims = 1:20)immune.combined <- FindClusters(immune.combined, resolution = 0.5)# Visualizationp1 <- DimPlot(immune.combined, reduction = "umap", group.by = "stim")p2 <- DimPlot(immune.combined, reduction = "umap", label = TRUE)plot_grid(p1, p2)

# 使用split.by参数根据不同的条件进行分割对比展示DimPlot(immune.combined, reduction = "umap", split.by = "stim")
鉴定保守的细胞类型标记基因
为了鉴定在不同处理下保守的细胞类型标记基因,我们提供了FindConservedMarkers功能函数。该函数会对不同数据集/组进行差异基因的表达分析,并使用MetaDE R包中的荟萃分析方法组合p值。
DefaultAssay(immune.combined) <- "RNA"# 使用FindConservedMarkers函数进行差异分析识别保守的细胞类型标记基因nk.markers <- FindConservedMarkers(immune.combined, ident.1 = 7, grouping.var = "stim", verbose = FALSE)head(nk.markers)## CTRL_p_val CTRL_avg_logFC CTRL_pct.1 CTRL_pct.2 CTRL_p_val_adj## SNHG12 1.059703e-193 1.3678805 0.335 0.019 1.489200e-189## HSPH1 2.552539e-139 2.0586178 0.553 0.100 3.587083e-135## NR4A2 8.671555e-136 1.5545769 0.296 0.023 1.218614e-131## SRSF2 1.556024e-113 1.6410606 0.704 0.220 2.186680e-109## BATF 1.573042e-09 0.5991204 0.116 0.042 2.210596e-05## CD69 1.188324e-122 1.8357378 0.525 0.096 1.669952e-118## STIM_p_val STIM_avg_logFC STIM_pct.1 STIM_pct.2 STIM_p_val_adj## SNHG12 8.090842e-159 1.054494 0.256 0.015 1.137006e-154## HSPH1 4.097380e-89 1.580183 0.471 0.114 5.758049e-85## NR4A2 3.130700e-78 1.009556 0.172 0.015 4.399572e-74## SRSF2 1.829674e-128 1.625081 0.675 0.182 2.571241e-124## BATF 9.234006e-126 1.354443 0.305 0.031 1.297655e-121## CD69 3.733167e-78 1.677068 0.688 0.291 5.246220e-74## max_pval minimump_p_val## SNHG12 8.090842e-159 2.119406e-193## HSPH1 4.097380e-89 5.105078e-139## NR4A2 3.130700e-78 1.734311e-135## SRSF2 1.556024e-113 3.659348e-128## BATF 1.573042e-09 1.846801e-125## CD69 3.733167e-78 2.376649e-122
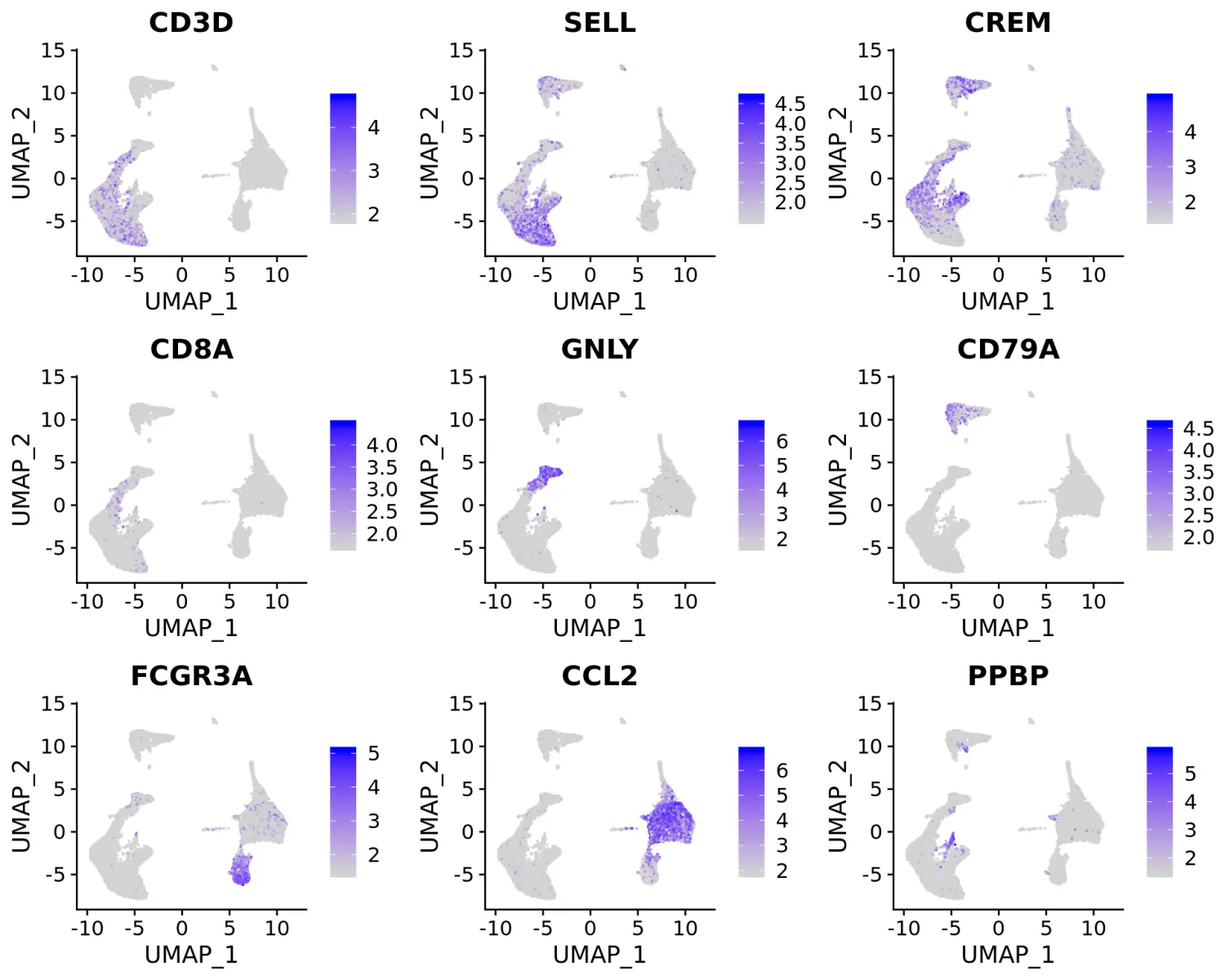
我们可以对这些marker基因进行可视化,并使用它们对不同的细胞类群进行注释。
# 可视化标记基因FeaturePlot(immune.combined, features = c("CD3D", "SELL", "CREM", "CD8A", "GNLY", "CD79A", "FCGR3A", "CCL2", "PPBP"), min.cutoff = "q9")
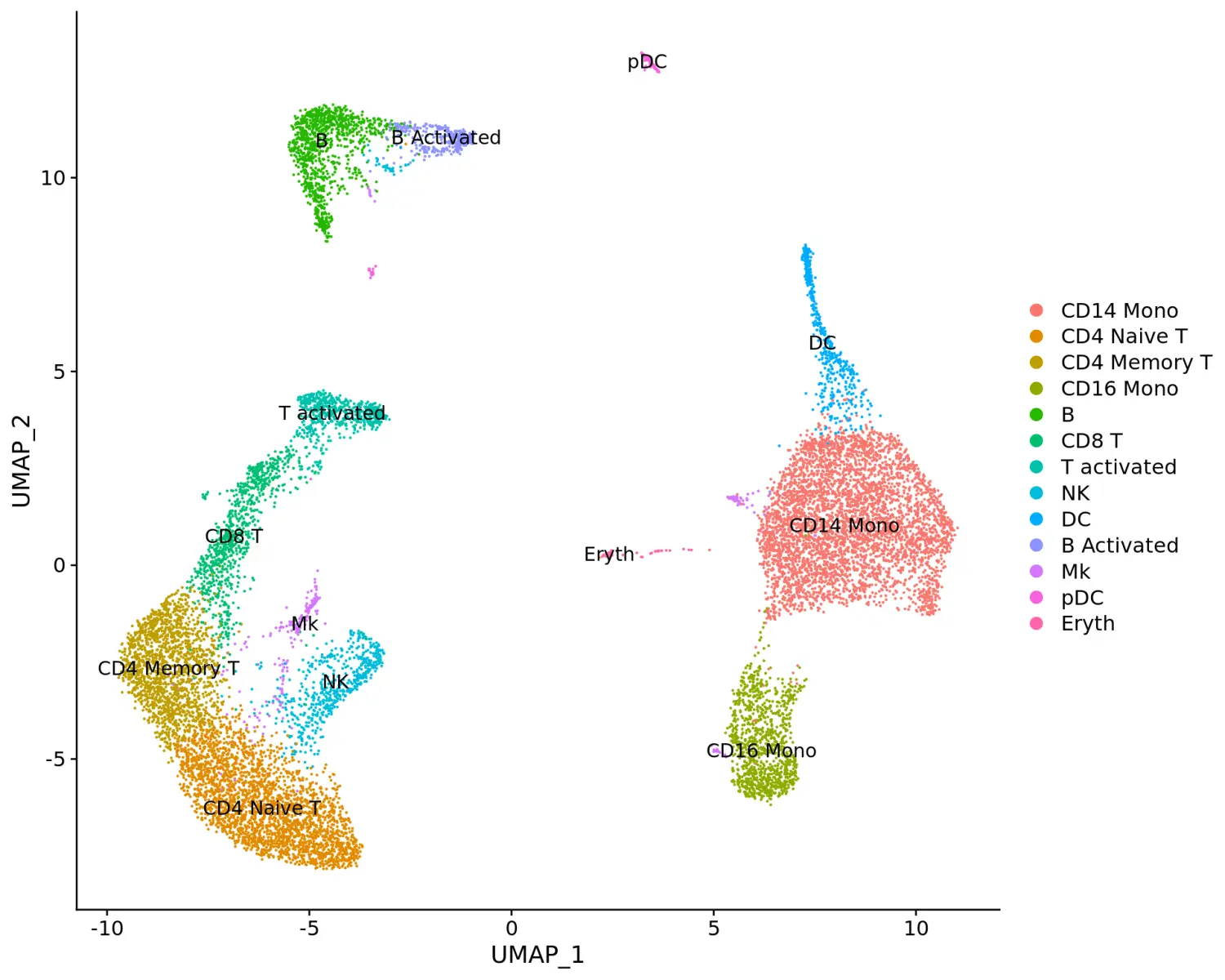
# 对不同细胞类群进行注释重命名immune.combined <- RenameIdents(immune.combined, `0` = "CD14 Mono", `1` = "CD4 Naive T", `2` = "CD4 Memory T", `3` = "CD16 Mono", `4` = "B", `5` = "CD8 T", `6` = "T activated", `7` = "NK", `8` = "DC", `9` = "B Activated", `10` = "Mk", `11` = "pDC", `12` = "Eryth", `13` = "Mono/Mk Doublets")# 细胞分群可视化DimPlot(immune.combined, label = TRUE)
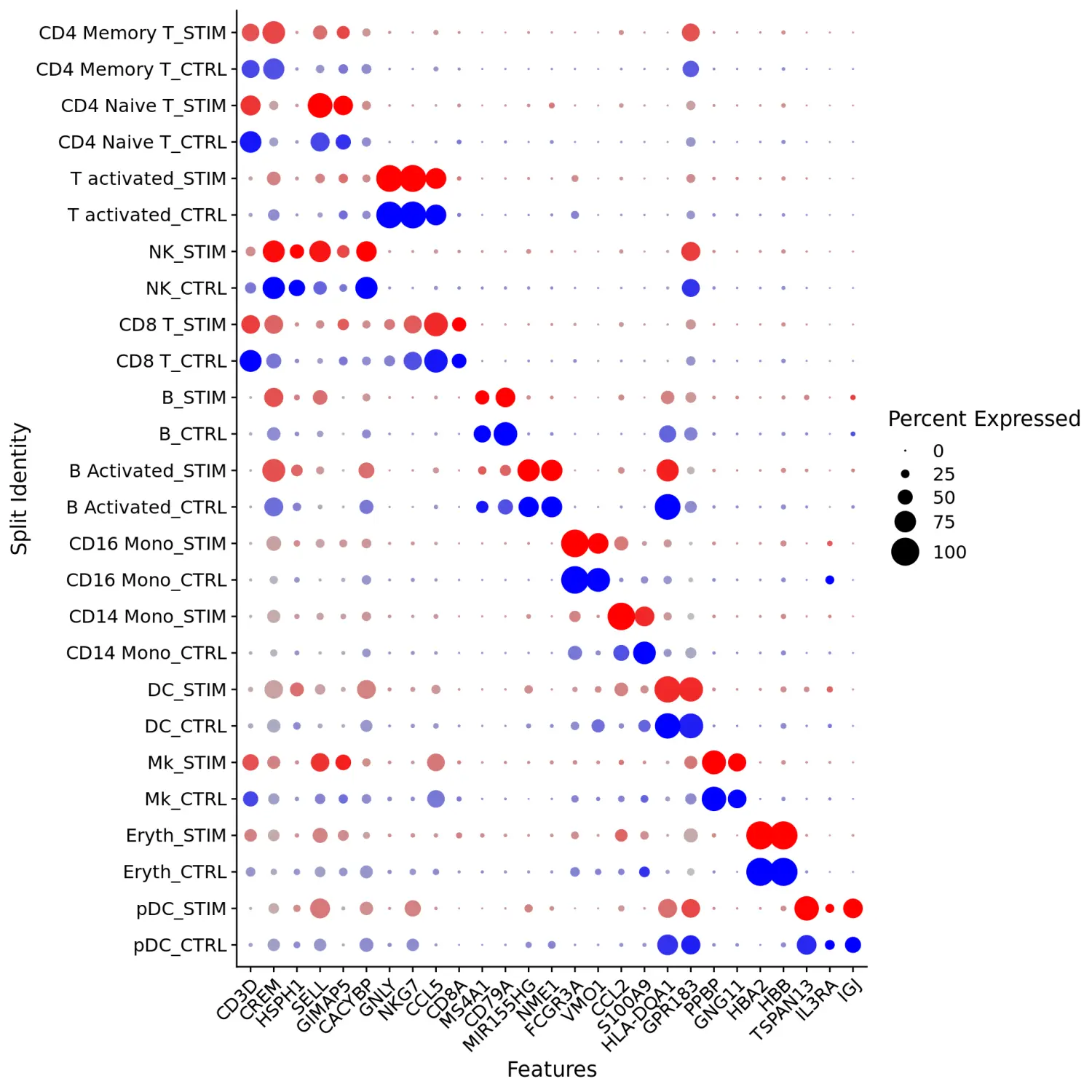
使用DotPlot函数对marker基因进行可视化,其中split.by参数可用于查看不同条件下的保守细胞类型marker基因,显示特定细胞类群中基因的表达水平和细胞的百分比。在这里,我们展示了13个细胞类群中每个群中的2-3个强标记基因。
Idents(immune.combined) <- factor(Idents(immune.combined), levels = c("Mono/Mk Doublets", "pDC", "Eryth", "Mk", "DC", "CD14 Mono", "CD16 Mono", "B Activated", "B", "CD8 T", "NK", "T activated", "CD4 Naive T", "CD4 Memory T"))markers.to.plot <- c("CD3D", "CREM", "HSPH1", "SELL", "GIMAP5", "CACYBP", "GNLY", "NKG7", "CCL5", "CD8A", "MS4A1", "CD79A", "MIR155HG", "NME1", "FCGR3A", "VMO1", "CCL2", "S100A9", "HLA-DQA1", "GPR183", "PPBP", "GNG11", "HBA2", "HBB", "TSPAN13", "IL3RA", "IGJ")DotPlot(immune.combined, features = rev(markers.to.plot), cols = c("blue", "red"), dot.scale = 8, split.by = "stim") + RotatedAxis()
鉴定不同刺激处理下的差异表达基因
接下来,我们将对不同刺激处理下的两组数据集进行比较分析。我们使用的方法是计算刺激组和对照组中细胞的平均表达,并绘制散点图识别那些异常表达的基因。在这里,我们计算了刺激组和对照组中naive T细胞和CD14单核细胞群体的平均表达并生成散点图,突出显示那些对干扰素刺激表现出强烈反应的基因。
library(ggplot2)library(cowplot)theme_set(theme_cowplot())# 提取特定细胞类群t.cells <- subset(immune.combined, idents = "CD4 Naive T")Idents(t.cells) <- "stim"# 计算平均表达水平并进行log转换avg.t.cells <- log1p(AverageExpression(t.cells, verbose = FALSE)$RNA)avg.t.cells$gene <- rownames(avg.t.cells)cd14.mono <- subset(immune.combined, idents = "CD14 Mono")Idents(cd14.mono) <- "stim"avg.cd14.mono <- log1p(AverageExpression(cd14.mono, verbose = FALSE)$RNA)avg.cd14.mono$gene <- rownames(avg.cd14.mono)# 选择特定基因进行突出标记genes.to.label = c("ISG15", "LY6E", "IFI6", "ISG20", "MX1", "IFIT2", "IFIT1", "CXCL10", "CCL8")p1 <- ggplot(avg.t.cells, aes(CTRL, STIM)) + geom_point() + ggtitle("CD4 Naive T Cells")p1 <- LabelPoints(plot = p1, points = genes.to.label, repel = TRUE)p2 <- ggplot(avg.cd14.mono, aes(CTRL, STIM)) + geom_point() + ggtitle("CD14 Monocytes")p2 <- LabelPoints(plot = p2, points = genes.to.label, repel = TRUE)plot_grid(p1, p2)

从散点图中我们可以看出,许多相同的基因在这两种细胞类型中的表达水平均被上调,这些基因可能代表了保守的干扰素应答途径。
immune.combined$celltype.stim <- paste(Idents(immune.combined), immune.combined$stim, sep = "_")immune.combined$celltype <- Idents(immune.combined)Idents(immune.combined) <- "celltype.stim"# 使用FindMarkers函数寻找差异表达基因b.interferon.response <- FindMarkers(immune.combined, ident.1 = "B_STIM", ident.2 = "B_CTRL", verbose = FALSE)head(b.interferon.response, n = 15)## p_val avg_logFC pct.1 pct.2 p_val_adj## ISG15 9.008631e-168 3.2061225 0.998 0.235 1.265983e-163## IFIT3 1.566158e-161 3.1240285 0.961 0.049 2.200921e-157## ISG20 1.206176e-158 2.0549983 1.000 0.662 1.695038e-154## IFI6 1.544804e-158 2.9139826 0.959 0.077 2.170914e-154## IFIT1 1.531805e-144 2.8529052 0.899 0.031 2.152645e-140## MX1 3.490304e-129 2.2712525 0.902 0.113 4.904924e-125## LY6E 3.320706e-127 2.1767293 0.897 0.146 4.666588e-123## TNFSF10 2.108526e-114 2.5996223 0.773 0.021 2.963111e-110## IFIT2 7.373988e-113 2.5114519 0.781 0.035 1.036267e-108## B2M 1.570724e-101 0.4194467 1.000 1.000 2.207338e-97## PLSCR1 8.204551e-101 1.9635626 0.792 0.115 1.152986e-96## IRF7 1.517587e-100 1.8203577 0.837 0.181 2.132665e-96## CXCL10 7.783861e-92 3.6869595 0.660 0.012 1.093866e-87## UBE2L6 1.659777e-88 1.4951634 0.855 0.296 2.332484e-84## EPSTI1 1.324448e-82 1.7540298 0.717 0.103 1.861247e-78
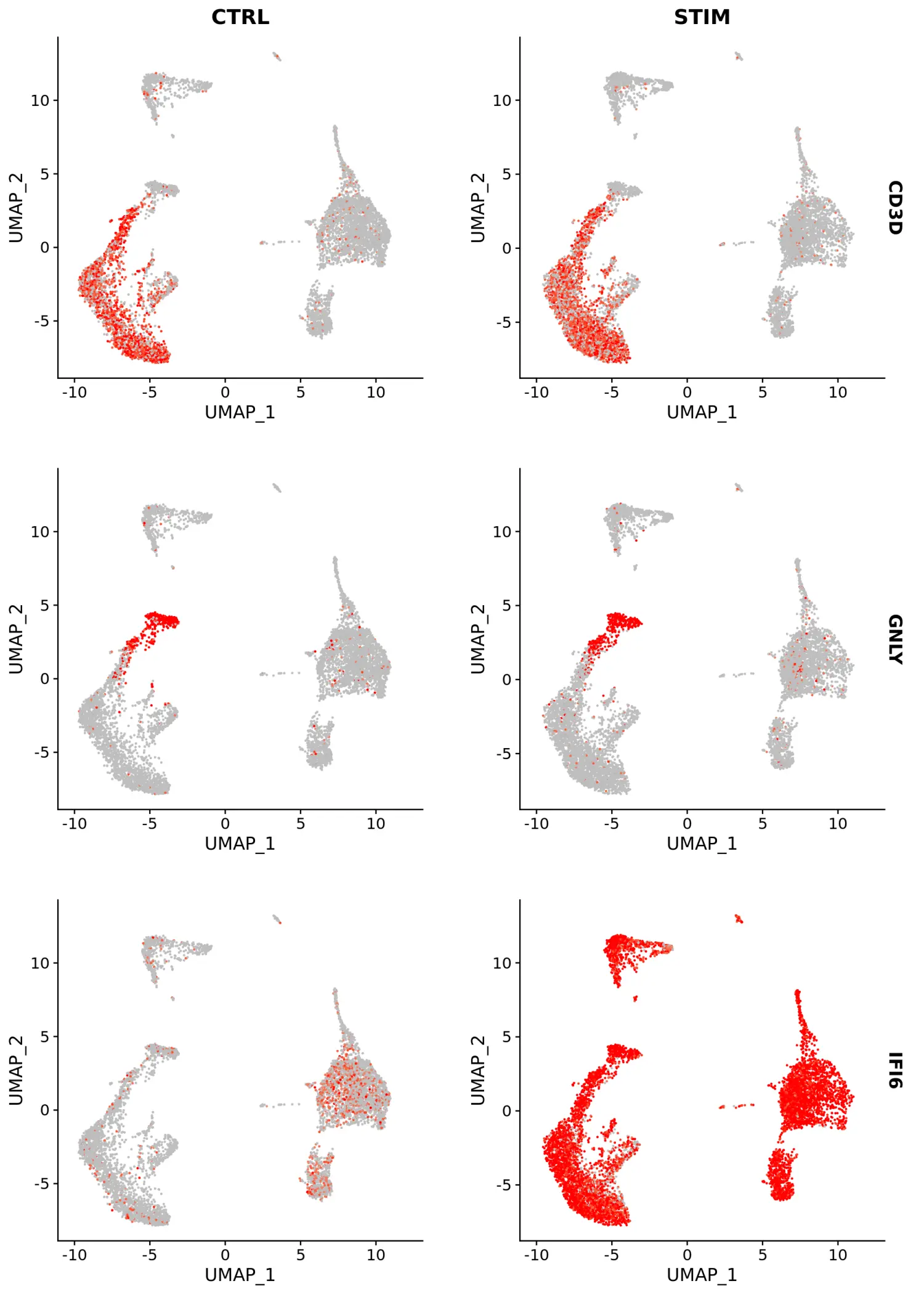
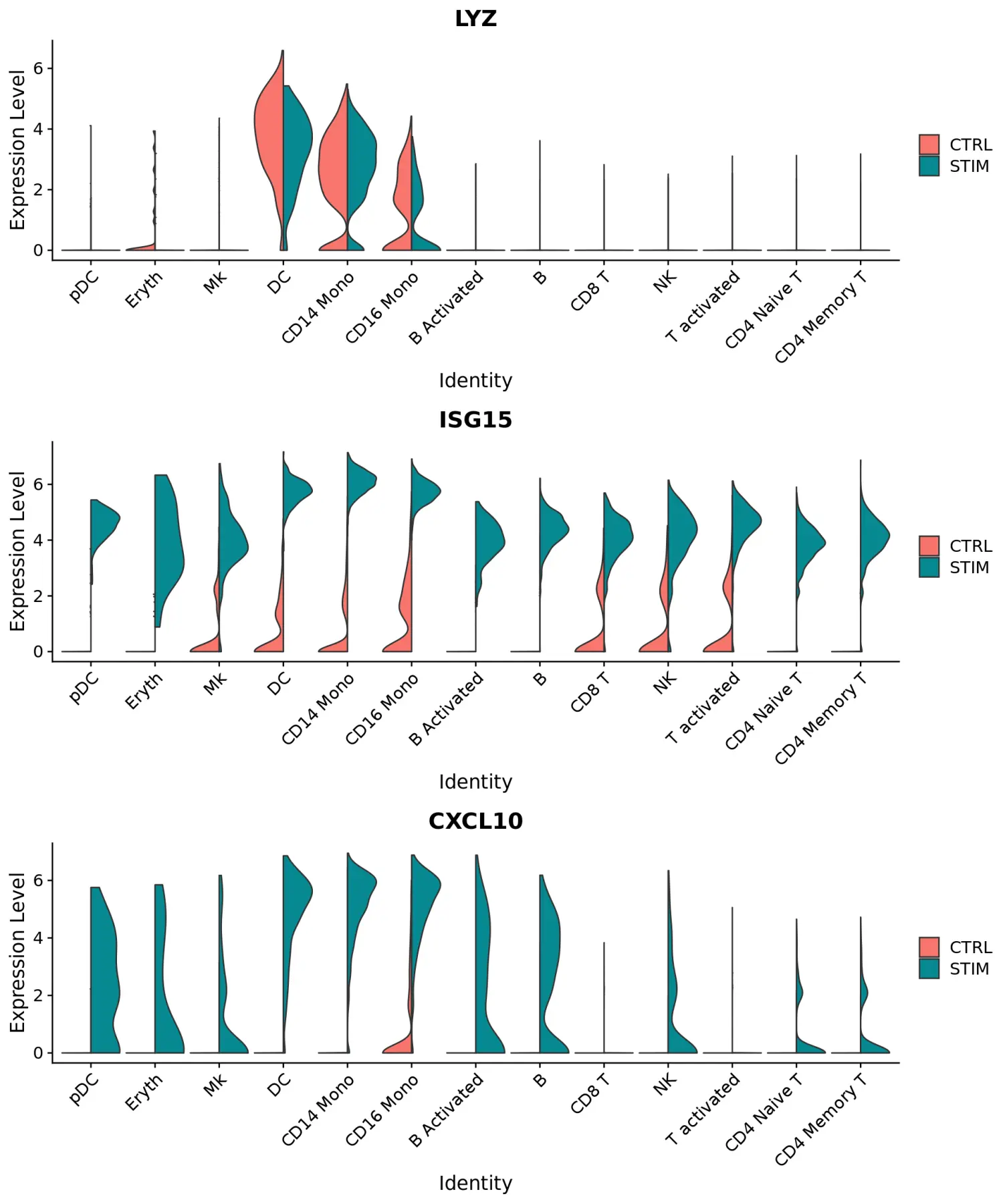
使用FeaturePlot和VlnPlot函数可视化标记基因,并设置split.by参数按分组变量(此处为刺激条件)进行划分。其中,CD3D和GNLY等基因是典型的细胞类型标记(对于T细胞和NK / CD8 T细胞),实际上不受干扰素刺激的影响,并且在对照组和受刺激组中显示出相似的基因表达模式。另一方面,IFI6和ISG15是核心的干扰素响应基因,因此在所有细胞类型中均被上调。最后,CD14和CXCL10是细胞类型特异性干扰素应答的基因。
FeaturePlot(immune.combined, features = c("CD3D", "GNLY", "IFI6"), split.by = "stim", max.cutoff = 3, cols = c("grey", "red"))
plots <- VlnPlot(immune.combined, features = c("LYZ", "ISG15", "CXCL10"), split.by = "stim", group.by = "celltype", pt.size = 0, combine = FALSE)CombinePlots(plots = plots, ncol = 1)
参考来源:https://satijalab.org/seurat/v3.1/immune_alignment.html

若有收获,就点个赞吧
0 人点赞
