Immunarch包也可以用于处理单细胞配对链数据,单细胞支持目前处于开发版本。为了访问它,我们需要通过执行以下命令来安装该软件包的最新开发版本:
install.packages ( "devtools" );devtools :: install_github ( "immunomind/immunarch" , ref = "dev" )
在Immunarch中,读取配对链免疫组库数据使用
repLoad函数并指定.mode = "paired"参数。目前我们仅支持10X Genomics。
我们可以使用select_barcodes函数通过特定barcode条形码对免疫组库进行子集化,也可以使用select_clusters函数选取创建特定聚类群或特定患者的数据集。
使用Immunarch包内置的单细胞配对链数据集
# 加载所需的R包library(immunarch)# 加载内置数据集data(scdata)# 查看数据信息names(scdata)# [1] "data" "meta" "bc_patient" "bc_cluster"head(scdata$meta)# A tibble: 1 x 1# Sample# <chr>#1 fluhead(scdata$bc_patient)# GCTGGGTTCAAACCGT-1 CAGTCCTCATGGTAGG-1 GGAAAGCGTCTTCAAG-1 CACAAACGTAAACGCG-1 CATGACATCCTCGCAT-1# "PatientA" "PatientA" "PatientA" "PatientA" "PatientA"# ATCCACCCAAGTCTGT-1# "PatientA"head(scdata$bc_cluster)#CAGTCCTTCGCGCCAA-1 TCGTAGATCTTCGGTC-1 CACACAAGTCAATACC-1 CGATGTAGTCGGCTCA-1 CTGTTTATCCTCTAGC-1# "Activ" "Activ" "Activ" "Activ" "Activ"# CACACTCCACGACTCG-1# "Activ"
加载配对链数据
要加载您自己的数据集,请使用该repLoad函数。目前,我们仅对 10X Genomics 数据实施了配对链数据支持。将数据集加载到 R 的工作示例:
file_path <- paste0(system.file(package = "immunarch"), "/extdata/sc/flu.csv.gz")igdata <- repLoad(file_path, .mode = "paired")## == Step 1/3: loading repertoire files... ==## Processing "<initial>" ...## -- Parsing "/private/var/folders/5d/g0z_tj9n3qd9r2b4tr8pp5sm0000gp/T/RtmpyOULE7/temp_libpath106e61c9ecf7b/immunarch/extdata/sc/flu.csv.gz" -- 10x (filt.contigs)## Warning: The following named parsers don't match the column names:## sample,barcode,is_cell,contig_id,high_confidence,length,chain,v_gene,d_gene,j_gene,c_gene,full_length,productive,cdr3,cdr3_nt,reads,umis,raw_clonotype_id,raw_consensus_id#### == Step 2/3: checking metadata files and merging files... ==#### Processing "<initial>" ...## -- Metadata file not found; creating a dummy metadata...#### == Step 3/3: processing paired chain data... ==#### Done!
查看加载成功后的数据信息
igdata$meta## # A tibble: 1 x 1## Sample## <chr>## 1 fluhead(igdata$data[[1]][c(1:7, 16, 17)])## Clones Proportion## 1 3 3e-04## 2 3 3e-04## 3 2 2e-04## 4 2 2e-04## 5 2 2e-04## 6 2 2e-04## CDR3.nt## 1 TGTGCACACACCACCGAACTCTATTGTACTAATGGTGTATGCTATGGGGGCTACTTTGACTACTGG;TGCCAACAGTATAATAGTTATTCGTGGACGTTC## 2 TGTGCGAGGCTATGGGGTTGGGGATTACTCTACTGG;TGCACCTCATATGCAGGCAGCAACAATTTGGTATTC## 3 TGTACCGCTCATGGTAGGGAGGGTACTTTTGATCTCTGG;TGTCAACAATATGATAATCTCCCTCGGTACACTTTT## 4 TGTGCAAGAGACGACTTTGCTTCGGGGGGTCGACACTTTGGCTGCTGG;TGTCAGCAGTCTGGTAACGCACCTCGAACTTTT## 5 TGTGCAAGAGATCTGGACTACATGGACGTCTGG;TGTCAACAGAGTTACAGTACCCCTCGAACTTTT## 6 TGTGCAAGAGGTTTAATACCCTCAGTTAGTGGCTACGACTACTACTACTACTACGGTATGGACGTCTGG;TGTCAATCAGCAGACAGCAGTGGTACTTATGAGGTATTC## CDR3.aa V.name D.name## 1 CAHTTELYCTNGVCYGGYFDYW;CQQYNSYSWTF IGHV2-5;IGKV1-5 IGHD2-8;None## 2 CARLWGWGLLYW;CTSYAGSNNLVF IGHV4-59;IGLV2-11 IGHD3-10;None## 3 CTAHGREGTFDLW;CQQYDNLPRYTF IGHV3-15;IGKV1-33 IGHD2-15;None## 4 CARDDFASGGRHFGCW;CQQSGNAPRTF IGHV1-2;IGKV3-20 IGHD3-10;None## 5 CARDLDYMDVW;CQQSYSTPRTF IGHV3-13;IGKV1D-39 None;None## 6 CARGLIPSVSGYDYYYYYGMDVW;CQSADSSGTYEVF IGHV3-74;IGLV3-25 IGHD5-12;None## J.name chain Barcode## 1 IGHJ4;IGKJ1 IGH;IGK AGTAGTCAGTGTACTC-1;GGCGACTGTACCGAGA-1;TTGAACGGTCACCTAA-1## 2 IGHJ4;IGLJ2 IGH;IGL AGAGCGACACCTTGTC-1;ATTGGTGAGACCTAGG-1;TCTTCGGAGGTGATTA-1## 3 IGHJ3;IGKJ2 IGH;IGK CGCGGTATCCTCTAGC-1;TGAGCATCAGGAACGT-1## 4 IGHJ4;IGKJ2 IGH;IGK ACTGTCCAGACGCAAC-1;CACACTCTCCGTTGTC-1## 5 IGHJ6;IGKJ2 IGH;IGK AGGTCCGAGTCAAGCG-1;GGCGTGTTCTCTAGGA-1## 6 IGHJ6;IGLJ2 IGH;IGL ACTGAGTCAAGCGATG-1;CTGCTGTCACGCATCG-1
提取barcode子集
要按barcode条形码对数据进行子集化,请使用该select_barcodes函数。
barcodes <- c("AGTAGTCAGTGTACTC-1", "GGCGACTGTACCGAGA-1", "TTGAACGGTCACCTAA-1")new_df <- select_barcodes(scdata$data[[1]], barcodes)new_df## CDR3.nt## 1 TGTGCACACACCACCGAACTCTATTGTACTAATGGTGTATGCTATGGGGGCTACTTTGACTACTGG;TGCCAACAGTATAATAGTTATTCGTGGACGTTC## CDR3.aa V.name D.name J.name V.end## 1 CAHTTELYCTNGVCYGGYFDYW;CQQYNSYSWTF IGHV2-5;IGKV1-5 NA IGHJ4;IGKJ1 NA## D.start D.end J.start VJ.ins VD.ins DJ.ins Sequence chain raw_clonotype_id## 1 NA NA NA NA NA NA NA IGH;IGK 14## ContigID Clones Barcode## 1 NA 3 AGTAGTCAGTGTACTC-1;GGCGACTGTACCGAGA-1;TTGAACGGTCACCTAA-1## Proportion## 1 1
提取特定病人的数据集
要创建具有特定病人免疫组库的新数据集,请使用以下select_clusters函数:
scdata_pat <- select_clusters(scdata, scdata$bc_patient, "Patient")names(scdata_pat$data)## [1] "flu_PatientA" "flu_PatientB" "flu_PatientC"scdata_pat$meta## # A tibble: 3 x 3## Sample Patient.source Patient## <chr> <chr> <chr>## 1 flu_PatientA flu PatientA## 2 flu_PatientB flu PatientB## 3 flu_PatientC flu PatientC
提取特定聚类群的数据集
要创建具有特定聚类群免疫组库的新数据集,请使用该select_clusters函数。您可以在创建特定于患者的数据集后应用此函数以获取特定于患者的细胞簇特定免疫组库,例如特定患者的记忆 B 细胞组库:
scdata_cl <- select_clusters(scdata_pat, scdata$bc_cluster, "Cluster")names(scdata_cl$data)## [1] "flu_PatientA_Activ" "flu_PatientA_Memory" "flu_PatientA_Naive"## [4] "flu_PatientB_Activ" "flu_PatientB_Memory" "flu_PatientB_Naive"## [7] "flu_PatientC_Activ" "flu_PatientC_Memory" "flu_PatientC_Naive"scdata_cl$meta## # A tibble: 9 x 5## Sample Patient.source Patient Cluster.source Cluster## <chr> <chr> <chr> <chr> <chr>## 1 flu_PatientA_Activ flu PatientA flu_PatientA Activ## 2 flu_PatientA_Memory flu PatientA flu_PatientA Memory## 3 flu_PatientA_Naive flu PatientA flu_PatientA Naive## 4 flu_PatientB_Activ flu PatientB flu_PatientB Activ## 5 flu_PatientB_Memory flu PatientB flu_PatientB Memory## 6 flu_PatientB_Naive flu PatientB flu_PatientB Naive## 7 flu_PatientC_Activ flu PatientC flu_PatientC Activ## 8 flu_PatientC_Memory flu PatientC flu_PatientC Memory## 9 flu_PatientC_Naive flu PatientC flu_PatientC Naive
探索和计算统计数据
对于单细胞配对链数据,immunarch包中的可视化函数可以同样的适用。
p1 <- repOverlap(scdata_cl$data) %>% vis()p2 <- repDiversity(scdata_cl$data) %>% vis()target <- c("CARAGYLRGFDYW;CQQYGSSPLTF", "CARATSFYYFHHW;CTSYTTRTTLIF", "CARDLSRGDYFPYFSYHMNVW;CQSDDTANHVIF", "CARGFDTNAFDIW;CTAWDDSLSGVVF", "CTREDYW;CMQTIQLRTF")p3 <- trackClonotypes(scdata_cl$data, target, .col = "aa") %>% vis()## Warning in melt.data.table(.data): id.vars and measure.vars are internally## guessed when both are 'NULL'. All non-numeric/integer/logical type columns are## considered id.vars, which in this case are columns [CDR3.aa]. Consider providing## at least one of 'id' or 'measure' vars in future.(p1 + p2) / p3
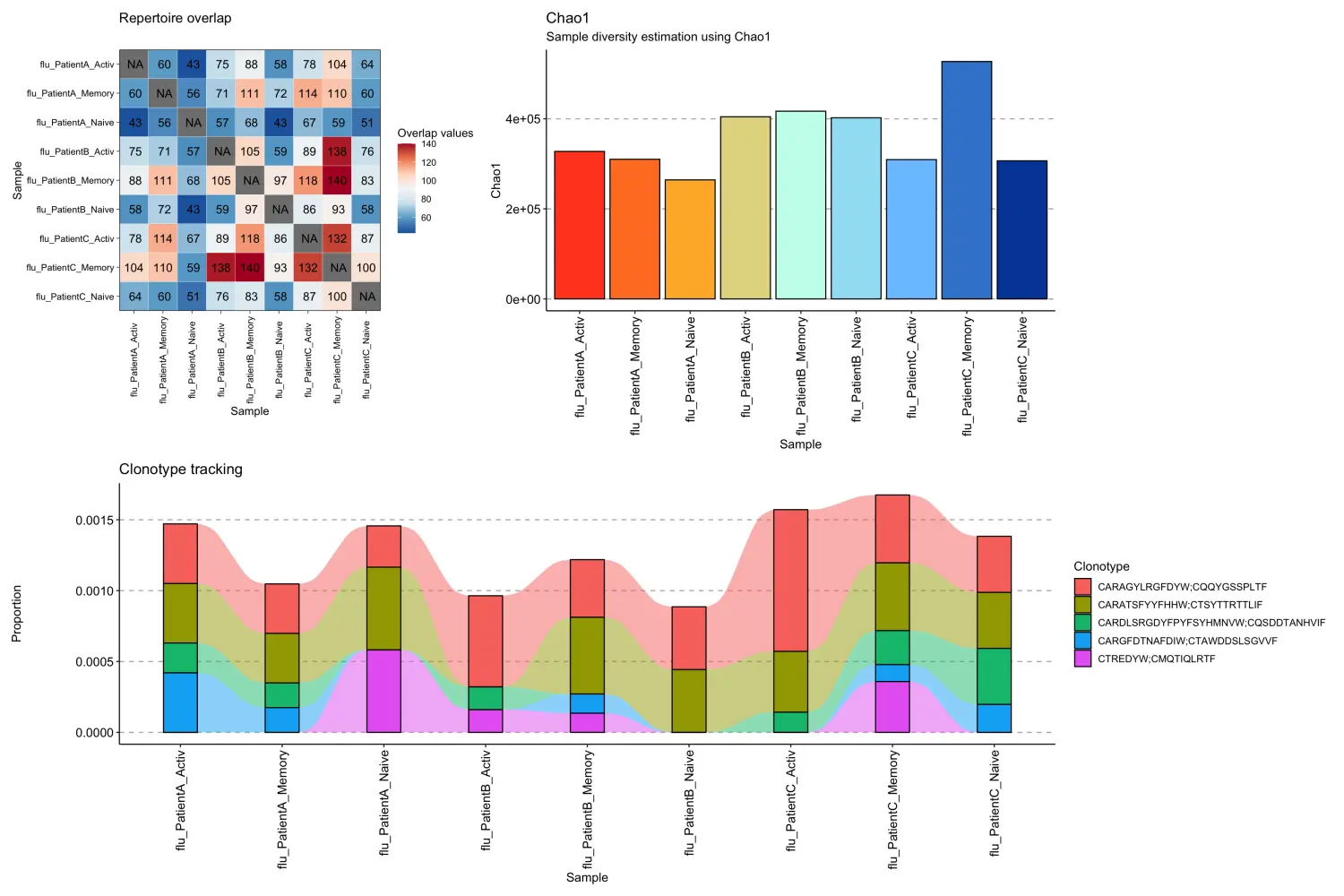
参考来源:https://immunarch.com/articles/web_only/v21_singlecell.html

若有收获,就点个赞吧
0 人点赞
