Monocle包是一个强大的单细胞转录组数据分析工具,它引入了一个对单细胞进行排序的新方法(拟时序分析),通过利用不同细胞转录调控的异步过程,将它们沿着与生物过程(如细胞分化等)相对应的发育轨迹进行排序。Monocle通过使用先进的机器学习算法(Reversed Graph Embedding反向图嵌入)从单细胞基因组数据中学习显式主图来对不同的单细胞进行排序,该方法对于解决那些复杂的生物学过程具有很高的鲁棒性和准确性。
Monocle2主要执行以下三种类型的分析:
- 对细胞进行聚类,分类和计数。
- 构建单细胞发育分化轨迹。
- 基因差异表达分析。
安装monocle2及其依赖包
source("http://bioconductor.org/biocLite.R")biocLite("monocle")biocLite(c("DDRTree", "pheatmap"))library(monocle)
monocle2基本分析流程
构建CellDataSet对象存储表达数据
# 加载样本的metadata注释信息pd <- new("AnnotatedDataFrame", data = sample_sheet)# 加载基因的metadata注释信息fd <- new("AnnotatedDataFrame", data = gene_annotation)# 使用newCellDataSet函数构建CellDataSet对象cds <- newCellDataSet(expr_matrix, phenoData = pd, featureData = fd)
通过marker基因对细胞进行分类
cth <- newCellTypeHierarchy()# 选择marker基因MYF5_id <- row.names(subset(fData(cds), gene_short_name == "MYF5"))ANPEP_id <- row.names(subset(fData(cds), gene_short_name == "ANPEP"))# 根据marker基因添加细胞类型注释cth <- addCellType(cth, "Myoblast", classify_func =function(x) { x[MYF5_id,] >= 1 })cth <- addCellType(cth, "Fibroblast", classify_func =function(x) { x[MYF5_id,] < 1 & x[ANPEP_id,] > 1 } )# 使用classifyCells函数根据marker基因对细胞进行分类cds <- classifyCells(cds, cth, 0.1)
对细胞进行聚类分群
# 使用clusterCells函数对细胞进行聚类分群cds <- clusterCells(cds)
拟时序分析对细胞进行排序
disp_table <- dispersionTable(cds)# 根据基因的表达选择排序的基因ordering_genes <- subset(disp_table, mean_expression >= 0.1)cds <- setOrderingFilter(cds, ordering_genes)# 使用reduceDimension函数进行数据降维cds <- reduceDimension(cds)# 使用orderCells函数对细胞进行排序cds <- orderCells(cds)
执行基因表达差异分析
# 使用differentialGeneTest函数进行差异分析diff_test_res <- differentialGeneTest(cds, fullModelFormulaStr = "~Media")sig_genes <- subset(diff_test_res, qval < 0.1)
数据导入与CellDataSet对象构建
Monocle2使用CellDataSet对象存储单细胞RNA-seq的基因表达矩阵及其相应的metadata注释信息,该对象衍生自Bioconductor中的ExpressionSet类。
CellDataSet类主要需要以下三个输入文件:
- exprs,表示数值的基因表达矩阵,其中行是基因,列是细胞。
- phenoData,一个AnnotatedDataFrame对象,其中行是细胞,列是细胞的属性(例如细胞的类型,培养条件,捕获的天数等)。
- featureData,一个AnnotatedDataFrame对象,其中行是特征(例如基因),列是基因的属性,例如生物型,gc含量等。
其中,这三个输入文件必须满足以下要求:
- 基因表达矩阵的列数与phenoData的行数相同。
- 基因表达矩阵的行数与featureData的行数相同。
- phenoData对象的行名称应与表达矩阵的列名称相匹配。
- featureData对象的行名应与表达矩阵的行名相匹配。
- featureData中必须有一个名为“gene_short_name”的列。
library(monocle)# 读取所需的三个输入文件# 基因表达矩阵HSMM_expr_matrix <- read.table("fpkm_matrix.txt")# 细胞注释信息HSMM_sample_sheet <- read.delim("cell_sample_sheet.txt")# 基因注释信息HSMM_gene_annotation <- read.delim("gene_annotations.txt")# 构建CellDataSet对象pd <- new("AnnotatedDataFrame", data = HSMM_sample_sheet)fd <- new("AnnotatedDataFrame", data = HSMM_gene_annotation)HSMM <- newCellDataSet(as.matrix(HSMM_expr_matrix),phenoData = pd, featureData = fd)
默认情况下,Monocle2假定我们读取的表达数据是以转录计数的原始count值,并使用负二项分布模型来进行下游的基因表达差异分析。
注意:如果我们有原始的UMI counts计数矩阵,则在创建CellDataSet对象之前不应该对其进行规归一化处理,也不应尝试将UMI计数转换为相对丰度(如将其转换为FPKM / TPM数据)。Monocle2将在内部执行所有必需的标准化步骤,自己提前进行归一化处理可能会破坏Monocle的一些关键步骤。
Monocle2也可以通过importCDS和exportCDS函数将其他格式的数据导入为CellDataSet对象,或将CellDataSet对象导出为其他格式。
# Where 'data_to_be_imported' can either be a Seurat object or an SCESet.# 将其他格式的数据导入为CellDataSet对象importCDS(data_to_be_imported)# We can set the parameter 'import_all' to TRUE if we'd like to import all the slots from our Seurat object or SCESet. (Default is FALSE or only keep minimal dataset)importCDS(data_to_be_imported, import_all = TRUE)lung <- load_lung()# To convert to Seurat object# 将CellDataSet对象导出为Seurat对象lung_seurat <- exportCDS(lung, 'Seurat')# To convert to SCESet# 将CellDataSet对象导出为SCESet对象lung_SCESet <- exportCDS(lung, 'Scater')
Choosing a distribution for your data
Monocle2既可以使用基因的相对表达丰度,也可以使用基于计数的原始count值作为输入的表达矩阵。通常,它最适合使用转录本的原始count值,尤其是UMI数据。无论我们使用哪种类型的数据,都必须为其指定适当的分布。FPKM / TPM值通常呈对数正态分布,而UMI或read counts可更好地使用负二项分布建模。对于原始的count计数数据,请将负二项式分布指定为newCellDataSet的expressionFamily参数。
#Do not runHSMM <- newCellDataSet(count_matrix,phenoData = pd,featureData = fd,expressionFamily=negbinomial.size())
以下是一些常用的expressionFamily值及其适用条件: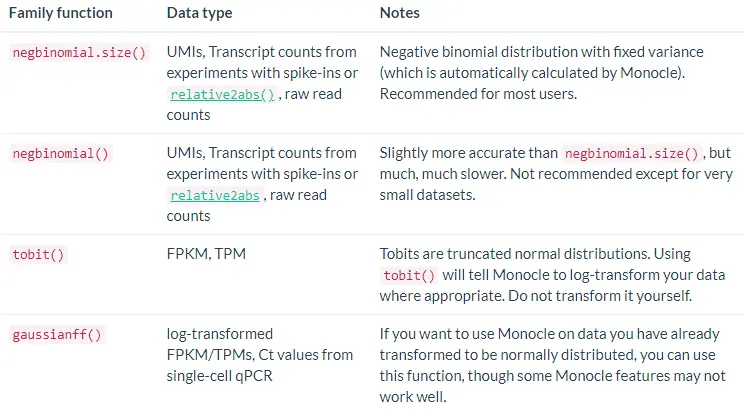
Notes:Using the
wrong expressionFamilyfor your data willlead to bad results, errors from Monocle, or both. However, if you have FPKM/TPM data, you canstill use negative binomialif you first convert your relative expression values to transcript counts usingrelative2abs(). This often leads to much more accurate results than usingtobit(). See the section onConverting TPM to mRNA Countsfor details.
Working with large data sets
对于一些大型的单细胞RNA-seq数据集,如具有50,000个细胞和25,000+个基因组成的基因表达矩阵会占用大量的内存。但是,由于当前的测序技术通常无法捕获每个细胞中的全部或大多数mRNA分子,因此许多基因的表达均会被检测为零。我们通常建议大多数用户使用稀疏矩阵sparseMatrices的表达数据,它会加快许多计算的速度。
我们可以使用Matrix包将基因表达矩阵转换为稀疏矩阵提供给Monocle:
HSMM <- newCellDataSet(as(umi_matrix, "sparseMatrix"),phenoData = pd,featureData = fd,lowerDetectionLimit = 0.5,expressionFamily = negbinomial.size())
如果我们有10X Genomics产生的counts数据,可以使用cellrangerRkit包中的load_cellranger_matrix函数直接加载基因表达矩阵。
cellranger_pipestance_path <- "/path/to/your/pipeline/output/directory"gbm <- load_cellranger_matrix(cellranger_pipestance_path)# 获取基因metadata注释信息fd <- fData(gbm)# The number 2 is picked arbitrarily in the line below.# Where "2" is placed you should place the column number that corresponds to your featureData's gene short names.# 将基因注释信息的第二列命名为gene_short_namecolnames(fd)[2] <- "gene_short_name"# 构建CellDataSet对象gbm_cds <- newCellDataSet(exprs(gbm),phenoData = new("AnnotatedDataFrame", data = pData(gbm)),featureData = new("AnnotatedDataFrame", data = fd),lowerDetectionLimit = 0.5,expressionFamily = negbinomial.size())
Converting TPM/FPKM values into mRNA counts
如果我们得到的基因表达矩阵是已经进行了归一化处理的数据(如FPKM或TPM值),我们可以使用relative2abs函数在创建CellDataSet对象之前将其转换为RPC值,如下所示:
pd <- new("AnnotatedDataFrame", data = HSMM_sample_sheet)fd <- new("AnnotatedDataFrame", data = HSMM_gene_annotation)# First create a CellDataSet from the relative expression levelsHSMM <- newCellDataSet(as.matrix(HSMM_expr_matrix),phenoData = pd,featureData = fd,lowerDetectionLimit = 0.1,expressionFamily = tobit(Lower = 0.1))# Next, use it to estimate RNA countsrpc_matrix <- relative2abs(HSMM, method = "num_genes")# Now, make a new CellDataSet using the RNA countsHSMM <- newCellDataSet(as(as.matrix(rpc_matrix), "sparseMatrix"),phenoData = pd,featureData = fd,lowerDetectionLimit = 0.5,expressionFamily = negbinomial.size())
变异系数计算和数据过滤预处理
Estimate size factors and dispersions
Monocle2使用estimateSizeFactors和estimateDispersions两个函数计算Size factors和”dispersion” values,Size factors可以帮助我们对不同细胞中mRNA表达的差异进行归一化处理,”dispersion” values将有助于后续的基因差异表达分析。
HSMM <- estimateSizeFactors(HSMM)HSMM <- estimateDispersions(HSMM)
Notes: estimateSizeFactors() and estimateDispersions() will only work, and are only needed, if you are working with a CellDataSet with a negbinomial() or negbinomial.size() expression family.
Filtering low-quality cells
Monocle2使用detectGenes函数查看基因的表达情况,并根据基因的表达预测其是否用于后续的细胞排序分析。
HSMM <- detectGenes(HSMM, min_expr = 0.1)print(head(fData(HSMM)))gene_short_name biotype num_cells_expressed use_for_orderingENSG00000000003.10 TSPAN6 protein_coding 184 FALSEENSG00000000005.5 TNMD protein_coding 0 FALSEENSG00000000419.8 DPM1 protein_coding 211 FALSEENSG00000000457.8 SCYL3 protein_coding 18 FALSEENSG00000000460.12 C1orf112 protein_coding 47 TRUEENSG00000000938.8 FGR protein_coding 0 FALSEprint(head(pData(HSMM)))Library Well Hours Media Mapped.Fragments Pseudotime State Size_Factor num_genes_expressedT0_CT_A01 SCC10013_A01 A01 0 GM 1958074 23.916673 1 1.392811 6850T0_CT_A03 SCC10013_A03 A03 0 GM 1930722 9.022265 1 1.311607 6947T0_CT_A05 SCC10013_A05 A05 0 GM 1452623 7.546608 1 1.218922 7019T0_CT_A06 SCC10013_A06 A06 0 GM 2566325 21.463948 1 1.013981 5560T0_CT_A07 SCC10013_A07 A07 0 GM 2383438 11.299806 1 1.085580 5998T0_CT_A08 SCC10013_A08 A08 0 GM 1472238 67.436042 2 1.099878 6055# 选取特定的基因expressed_genes <- row.names(subset(fData(HSMM),num_cells_expressed >= 50))
现在,expressed_genes向量中存储着至少在50个细胞中表达的基因,我们将用其对后续的细胞按照生物学过程的顺序进行排序的分析。
# 对细胞进行过滤valid_cells <- row.names(subset(pData(HSMM),Cells.in.Well == 1 &Control == FALSE &Clump == FALSE &Debris == FALSE &Mapped.Fragments > 1000000))HSMM <- HSMM[,valid_cells]# 根据基因的表达对基因进行过滤pData(HSMM)$Total_mRNAs <- Matrix::colSums(exprs(HSMM))HSMM <- HSMM[,pData(HSMM)$Total_mRNAs < 1e6]upper_bound <- 10^(mean(log10(pData(HSMM)$Total_mRNAs)) +2*sd(log10(pData(HSMM)$Total_mRNAs)))lower_bound <- 10^(mean(log10(pData(HSMM)$Total_mRNAs)) -2*sd(log10(pData(HSMM)$Total_mRNAs)))# 查看基因的表达分布qplot(Total_mRNAs, data = pData(HSMM), color = Hours, geom ="density") +geom_vline(xintercept = lower_bound) +geom_vline(xintercept = upper_bound)
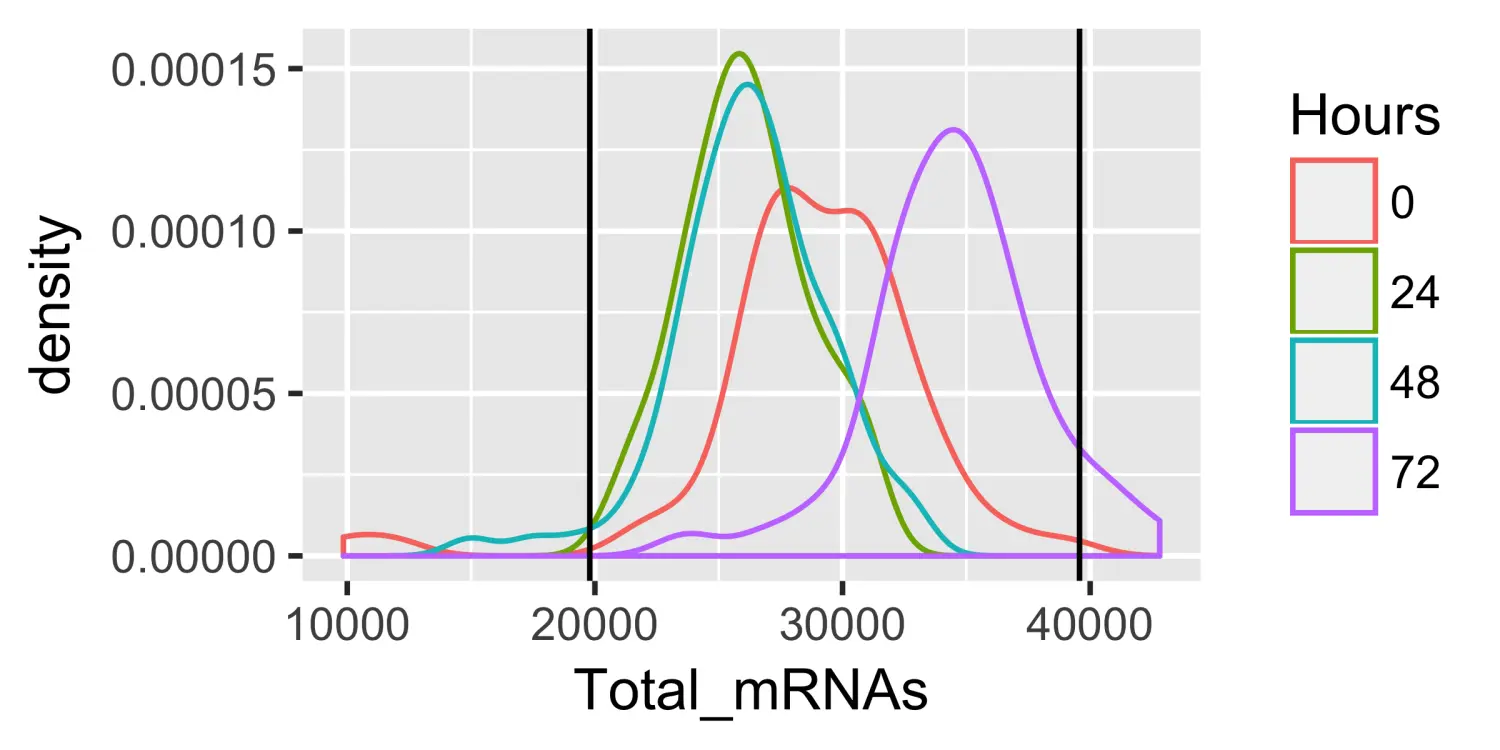
# 过滤掉那些极高表达和极低表达的细胞HSMM <- HSMM[,pData(HSMM)$Total_mRNAs > lower_bound &pData(HSMM)$Total_mRNAs < upper_bound]HSMM <- detectGenes(HSMM, min_expr = 0.1)
# Log-transform each value in the expression matrix.L <- log(exprs(HSMM[expressed_genes,]))# Standardize each gene, so that they are all on the same scale, Then melt the data with plyr so we can plot it easilymelted_dens_df <- melt(Matrix::t(scale(Matrix::t(L))))# Plot the distribution of the standardized gene expression values.qplot(value, geom = "density", data = melted_dens_df) +stat_function(fun = dnorm, size = 0.5, color = 'red') +xlab("Standardized log(FPKM)") +ylab("Density")

参考来源:http://cole-trapnell-lab.github.io/monocle-release/docs/



若有收获,就点个赞吧
0 人点赞
