免疫组库重叠分析
免疫组库重叠(Repertoire overlap)是度量不同样本之间TCR或BCR库相似性的最常用方法。它是通过计算在给定的Repertoire之间共享的克隆型(也称为“公共”克隆型)的特定统计数据来实现的。immunarch提供了几个指标: - 公共克隆型的数量 ( .method = "public") - 一个重叠相似性的经典度量指标。
- overlap coefficient,重叠系数 (
.method = "overlap") - 重叠相似性的标准化度量。它被定义为交集的大小除以两个集合中的较小者。 - Jaccard index,Jaccard指数 (
.method = "jaccard") - 它衡量有限样本集之间的相似度,定义为交集的大小除以样本集并集的大小。 - Tversky index,Tversky指数 (
.method = "tversky") - 一种集合上的非对称相似性度量,它将变体与原型进行比较。如果使用默认参数,则类似于 Dice 的系数。 - cosine similarity,余弦相似度 (
.method = "cosine") - 两个非零向量之间相似度的度量 - Morisita’s overlap index,Morisita重叠指数 (
.method = "morisita") - 一种用于计算个体在总体中的分散程度的统计量度。它用于比较样本之间的重叠。 - incremental overlap,增量重叠 - 计算N个最丰富的克隆型与增量增长的N的重叠(
.method = "inc+METHOD"例如,"inc+public"或"inc+morisita")。
我们可以使用repOverlap函数计算不同样本之间Repertoire的重叠情况。同样,我们可以将分析结果传递给vis()函数,以完成所有输出结果的可视化展示。
library(immunarch) # Load the package into Rdata(immdata) # Load the test dataset# 使用不同的度量方法计算Repertoire的重叠情况imm_ov1 <- repOverlap(immdata$data, .method = "public", .verbose = F)head(imm_ov1)# A2-i129 A2-i131 A2-i133 A2-i132 A4-i191 A4-i192 MS1 MS2 MS3 MS4 MS5 MS6#A2-i129 NA 63 74 69 46 55 30 41 24 35 44 54#A2-i131 63 NA 56 81 42 64 34 31 33 33 23 49#A2-i133 74 56 NA 87 49 61 41 44 31 31 44 65#A2-i132 69 81 87 NA 62 67 47 46 50 48 60 76#A4-i191 46 42 49 62 NA 55 42 34 41 29 37 49#A4-i192 55 64 61 67 55 NA 56 37 27 37 56 61imm_ov2 <- repOverlap(immdata$data, .method = "morisita", .verbose = F)head(imm_ov2)# A2-i129 A2-i131 A2-i133 A2-i132 A4-i191 A4-i192#A2-i129 NA 0.0024642881 0.0011511984 0.0044505612 0.0005804524 0.0024253356#A2-i131 0.0024642881 NA 0.0011475178 0.0088347844 0.0006212924 0.0019547325#A2-i133 0.0011511984 0.0011475178 NA 0.0043090343 0.0004456898 0.0016076124#A2-i132 0.0044505612 0.0088347844 0.0043090343 NA 0.0009178361 0.0023583418#A4-i191 0.0005804524 0.0006212924 0.0004456898 0.0009178361 NA 0.0005006889#A4-i192 0.0024253356 0.0019547325 0.0016076124 0.0023583418 0.0005006889 NA# MS1 MS2 MS3 MS4 MS5 MS6#A2-i129 0.0003009428 0.0011482287 0.0001797280 0.0014031280 0.0007196454 0.0027679140#A2-i131 0.0001927309 0.0014283644 0.0002328510 0.0021404462 0.0002198598 0.0034297172#A2-i133 0.0002194163 0.0018138252 0.0001618185 0.0007751521 0.0002272166 0.0017382456#A2-i132 0.0004486568 0.0032894737 0.0005874910 0.0073378655 0.0008173229 0.0106015902#A4-i191 0.0007469433 0.0002730513 0.0001892369 0.0004114056 0.0003530021 0.0008469919#A4-i192 0.0002977945 0.0007443917 0.0001358868 0.0016537104 0.0003422629 0.0544339382p1 <- vis(imm_ov1)p2 <- vis(imm_ov2, .text.size = 2)p1 + p2
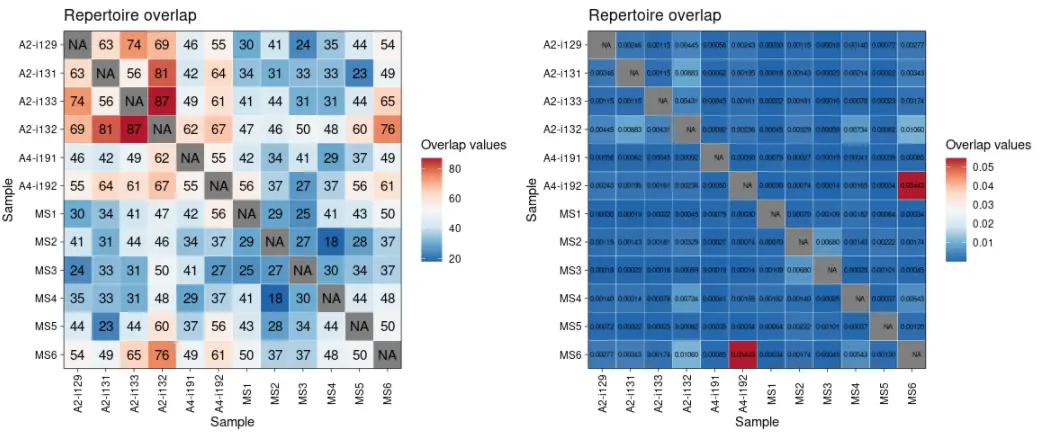
vis(imm_ov1, "heatmap2")
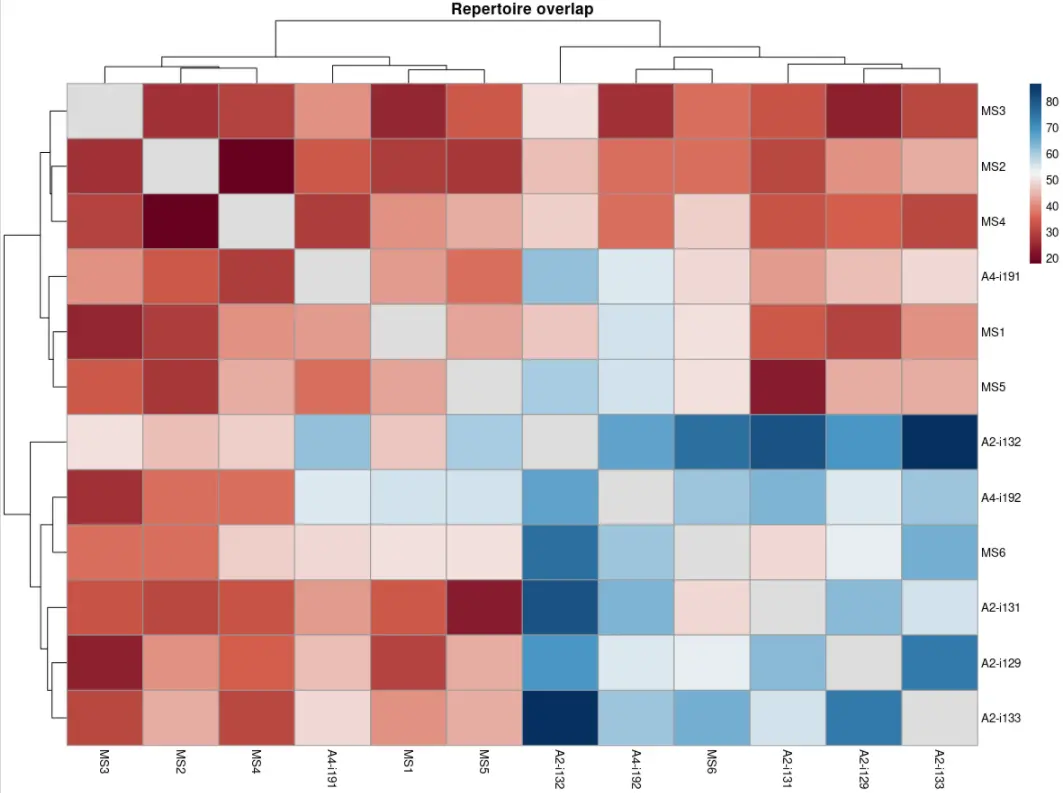
我们可以设置更改有效数字的位数:
p1 <- vis ( imm_ov2 , .text.size = 2.5 , .signif.digits = 1 )p2 <- vis ( imm_ov2 , .text.size = 2 , .signif.digits = 2 )p1 + p2
我们还可以使用repOverlapAnalysis函数对计算得到的重叠指标进行分析。
# Apply different analysis algorithms to the matrix of public clonotypes:# "mds" - Multi-dimensional Scaling# MDS降维repOverlapAnalysis(imm_ov1, "mds")## Standard deviations (1, .., p=4):## [1] 0 0 0 0#### Rotation (n x k) = (12 x 2):## [,1] [,2]## A2-i129 -18.7767715 -18.360817## A2-i131 29.9586985 -7.870441## A2-i133 28.1148594 22.629093## A2-i132 -44.3435640 6.221812## A4-i191 13.8586515 7.452149## A4-i192 -14.0065477 27.068830## MS1 -8.8469009 -8.151574## MS2 -0.9712073 -1.297017## MS3 -10.4398629 4.894354## MS4 0.5131505 10.471309## MS5 18.5153823 -12.628029## MS6 6.4241122 -30.429669
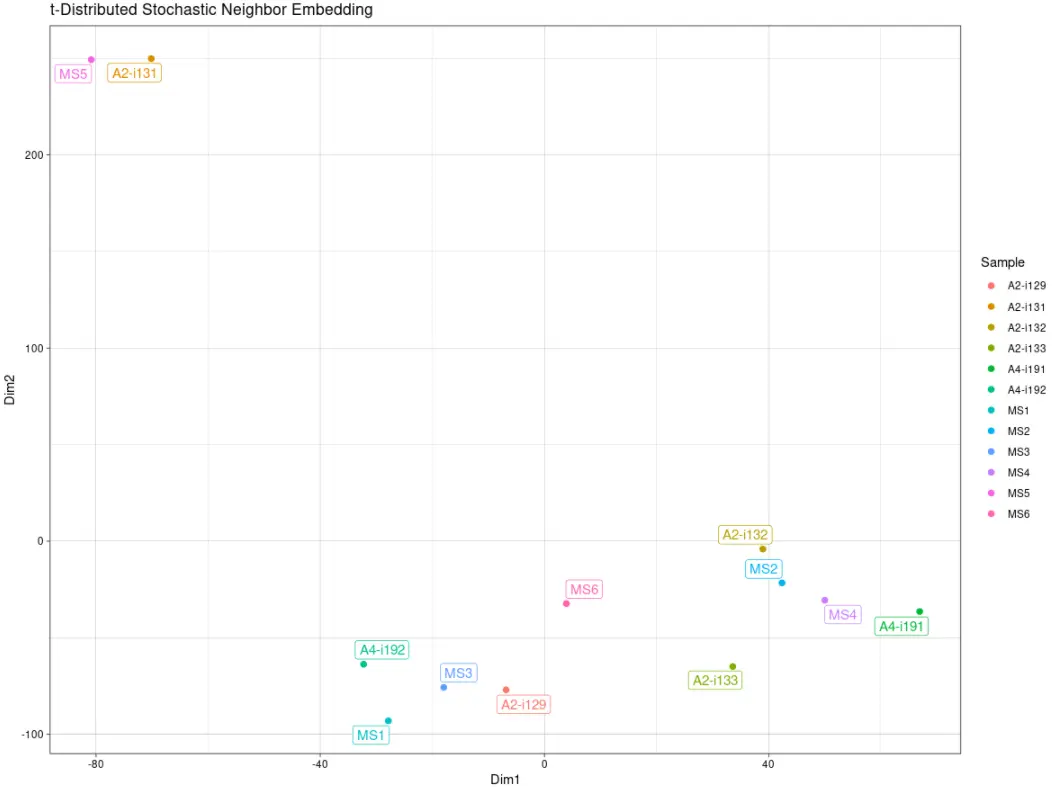
# "tsne" - t-Stochastic Neighbor Embedding# TSNE降维repOverlapAnalysis(imm_ov1, "tsne")## DimI DimII## A2-i129 141.757274 -1.875981## A2-i131 -336.372028 177.099380## A2-i133 82.395447 -42.878936## A2-i132 -11.731661 -41.878646## A4-i191 38.681498 -109.060994## A4-i192 169.797839 36.512757## MS1 116.225222 42.689482## MS2 3.659358 -70.354712## MS3 139.036548 18.337403## MS4 21.703642 -81.842537## MS5 -320.584081 165.745570## MS6 -44.569057 -92.492786## attr(,"class")## [1] "immunr_tsne" "matrix"
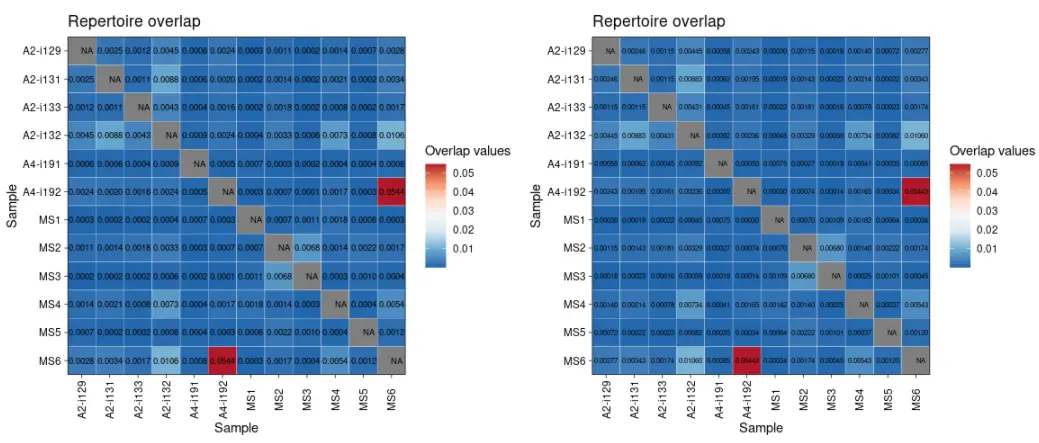
# Visualise the results# MDS降维可视化repOverlapAnalysis(imm_ov1, "mds") %>% vis()

# Visualise the results# TSNE降维可视化repOverlapAnalysis(imm_ov1, "tsne") %>% vis()
# Clusterise the MDS resulting components using K-meansrepOverlapAnalysis(imm_ov1, "mds+kmeans") %>% vis()

构建公共克隆型库
为了从repertoires列表中构建一个包含所有clonotypes的大型公共克隆型库,我们可以使用pubRep该函数。
# Pass "nt" as the second parameter to build the public repertoire table using CDR3 nucleotide sequences# 使用CDR3区域的核苷酸序列计算构建公共克隆型库pr.nt <- pubRep(immdata$data, "nt", .verbose = F)pr.nt## CDR3.nt Samples A2-i129## 1: TGCGCCAGCAGCTTGGAAGAGACCCAGTACTTC 8 1## 2: TGTGCCAGCAGCTTCCAAGAGACCCAGTACTTC 7 NA## 3: TGTGCCAGCAGTTACCAAGAGACCCAGTACTTC 7 1## 4: TGCGCCAGCAGCTTCCAAGAGACCCAGTACTTC 6 2## 5: TGTGCCAGCAGCCAAGAGACCCAGTACTTC 6 4## ---## 75101: TGTGCTTCACAACTCTTATTGGACGAGACCCAGTACTTC 1 NA## 75102: TGTGCTTCACAAGCCCTACAGGGCACTTTCCATAATTCACCCCTCCACTTT 1 NA## 75103: TGTGCTTCAGGGCGGGCCTACGAGCAGTACTTC 1 NA## 75104: TGTGCTTCCGCCGGACCGGACCGGGAGACCCAGTACTTC 1 NA## 75105: TGTGCTTGCGGGACAGATAACTATGGCTACACCTTC 1 NA## A2-i131 A2-i133 A2-i132 A4-i191 A4-i192 MS1 MS2 MS3 MS4 MS5 MS6## 1: NA 1 1 NA 1 NA NA 1 1 1 1## 2: 1 1 2 1 NA 1 NA NA 2 NA 1## 3: 1 1 NA 1 1 1 NA 2 NA NA NA## 4: NA 1 1 NA NA NA 1 NA 1 NA 1## 5: 2 NA 2 3 1 NA NA NA NA 4 NA## ---## 75101: 1 NA NA NA NA NA NA NA NA NA NA## 75102: NA NA NA NA NA NA NA NA NA 1 NA## 75103: NA NA NA NA NA 1 NA NA NA NA NA## 75104: NA 1 NA NA NA NA NA NA NA NA NA## 75105: NA NA NA NA 1 NA NA NA NA NA NA
# Pass "aa+v" as the second parameter to build the public repertoire table using CDR3 aminoacid sequences and V alleles# In order to use only CDR3 aminoacid sequences, just pass "aa"# 使用CDR3区域的氨基酸序列和V等位基因序列计算构建公共克隆型库pr.aav <- pubRep(immdata$data, "aa+v", .verbose = F)pr.aav## CDR3.aa V.name Samples A2-i129 A2-i131 A2-i133 A2-i132## 1: CASSLEETQYF TRBV5-1 8 1 NA 2 1## 2: CASSDSSGGANEQFF TRBV6-4 6 1 1 2 NA## 3: CASSFQETQYF TRBV5-1 6 3 NA 1 1## 4: CASSLGETQYF TRBV12-4 6 2 NA NA 4## 5: CASSDSGGSYNEQFF TRBV6-4 5 NA NA NA 3## ---## 74440: CTSSRPTQGAYEQYF TRBV7-2 1 NA NA NA NA## 74441: CTSSSRAGAGTDTQYF TRBV7-2 1 NA NA NA NA## 74442: CTSSYPGLAGLKRKETQYF TRBV7-2 1 NA NA NA 1## 74443: CTSSYRQRPYQETQYF TRBV7-2 1 NA NA NA NA## 74444: CTSSYSTSGVGQFF TRBV7-2 1 NA NA NA NA## A4-i191 A4-i192 MS1 MS2 MS3 MS4 MS5 MS6## 1: NA 2 NA NA 1 1 1 1## 2: 3 NA NA NA 2 NA NA 12## 3: NA NA NA 1 NA 1 NA 1## 4: 3 NA 1 NA NA NA 2 1## 5: NA 1 1 NA 1 NA NA 1## ---## 74440: NA NA NA NA NA NA NA 1## 74441: NA NA NA NA 1 NA NA NA## 74442: NA NA NA NA NA NA NA NA## 74443: NA NA NA NA 1 NA NA NA## 74444: NA NA NA NA NA 1 NA NA
# You can also pass the ".coding" parameter to filter out all noncoding sequences first:# 也可以通过设置“.coding=T”参数,事先过滤掉所有非编码序列:pr.aav.cod <- pubRep(immdata$data, "aa+v", .coding = T)
# Create a public repertoire with coding-only sequences using both CDR3 amino acid sequences and V genes# 使用 CDR3 氨基酸序列和 V 基因创建一个仅包含编码序列的公共库pr <- pubRep(immdata$data, "aa+v", .coding = T, .verbose = F)head(pr)# CDR3.aa V.name Samples A2-i129 A2-i131 A2-i133 A2-i132 A4-i191 A4-i192 MS1 MS2 MS3#1: CASSLEETQYF TRBV5-1 8 1 NA 2 1 NA 2 NA NA 1#2: CASSDSSGGANEQFF TRBV6-4 6 1 1 2 NA 3 NA NA NA 2#3: CASSFQETQYF TRBV5-1 6 3 NA 1 1 NA NA NA 1 NA#4: CASSLGETQYF TRBV12-4 6 2 NA NA 4 3 NA 1 NA NA#5: CASSDSGGSYNEQFF TRBV6-4 5 NA NA NA 3 NA 1 1 NA 1#6: CASSDSSGSTDTQYF TRBV6-4 5 NA NA NA 4 1 1 NA NA 1# MS4 MS5 MS6#1: 1 1 1#2: NA NA 12#3: 1 NA 1#4: NA 2 1#5: NA NA 1#6: NA NA 2# Apply the filter subroutine to leave clonotypes presented only in healthy individuals# 应用过滤子程序,过滤出只出现在健康个体中克隆型pr1 <- pubRepFilter(pr, immdata$meta, .by = c(Status = "C"))# Apply the filter subroutine to leave clonotypes presented only in diseased individuals# 应用过滤子程序,过滤出只出现在患病个体中克隆型pr2 <- pubRepFilter(pr, immdata$meta, .by = c(Status = "MS"))# Divide one by anotherpr3 <- pubRepApply(pr1, pr2)# Plot itp <- ggplot() +geom_jitter(aes(x = "Treatment", y = Result), data = pr3)p
参考来源:https://immunarch.com/articles/web_only/v4_overlap.html

若有收获,就点个赞吧
0 人点赞
