由於語法渲染問題而影響閱讀體驗, 請移步博客閱讀~
本文GitPage地址
Sanger Sequencing Plot (Biopython + Matplotlib)
Quick Start
from Bio import SeqIOimport matplotlib.pyplot as pltfrom collections import defaultdict## Abi Fileabi_file = "D10.1.F.YP13033649.D10.S7695.ab1"record = SeqIO.read(abi_file, "abi")channels = ["DATA1", "DATA2", "DATA3", "DATA4"]trace = defaultdict(list)for c in channels:trace[c] = record.annotations["abif_raw"][c]plt.plot(trace["DATA2"], color="green" ,alpha=0.6, lw=0.2) # Aplt.plot(trace["DATA4"], color="blue" ,alpha=0.6, lw=0.2) # Cplt.plot(trace["DATA1"], color="black" ,alpha=0.6, lw=0.2) # Gplt.plot(trace["DATA3"], color="red" ,alpha=0.6, lw=0.2) # Tplt.title(record.annotations['abif_raw']['TUBE1'])plt.show()
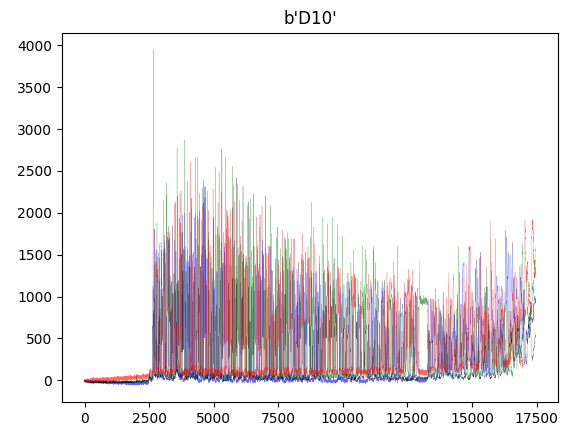
Script for Results of 96-wells plate
abi_plot.py
##!/usr/bin/env python3import argparseparser = argparse.ArgumentParser()parser.add_argument('-i','-I','--input') #输入文件parser.add_argument('-o','-O','--output', default="out.png") #输入文件##获取参数args = parser.parse_args()INPUT = args.inputOUTPUT = args.output##########import osimport matplotlib as mplimport matplotlib.pyplot as pltfrom Bio import SeqIOfrom collections import defaultdictdef raw_plot(INPUT):record = SeqIO.read(INPUT, "abi")channels = ["DATA1", "DATA2", "DATA3", "DATA4"]trace = defaultdict(list)for c in channels:trace[c] = record.annotations["abif_raw"][c]plt.plot(trace["DATA2"], color="green" ,alpha=0.6, lw=0.2) # Aplt.plot(trace["DATA4"], color="blue" ,alpha=0.6, lw=0.2) # Cplt.plot(trace["DATA1"], color="black" ,alpha=0.6, lw=0.2) # Gplt.plot(trace["DATA3"], color="red" ,alpha=0.6, lw=0.2) # Tplt.title(record.annotations['abif_raw']['TUBE1'])#plt.show()Cmd = "ls "+ str(INPUT)LIST = os.popen(Cmd).read().split("\n")[:-1]print(LIST)plt.figure(figsize=(14*3, 8*3))plt.ion()for i in range(96):plt.subplot(8,12,i+1)abi = INPUT+"/"+LIST[i]print(abi)raw_plot(abi)plt.show()plt.savefig(OUTPUT)
Usage:
abi_plot.py -i 96Well_Result
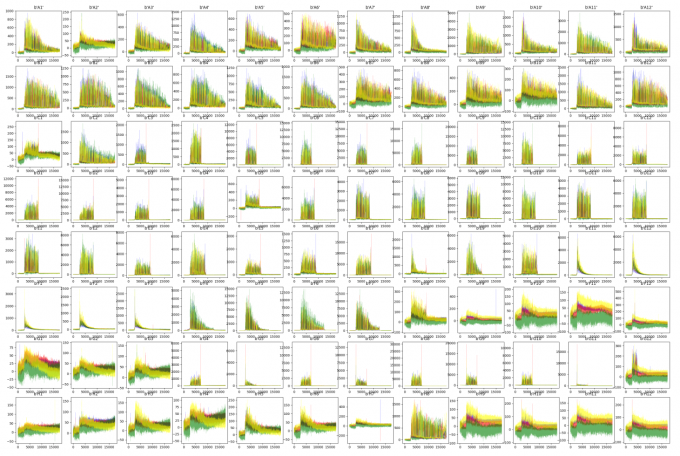
Box plot
It is useful when all abi files are plasmid or long sequencing result.
Box plot
def raw_box_plot(INPUT):record = SeqIO.read(INPUT, "abi")channels = ["DATA1", "DATA2", "DATA3", "DATA4"]trace = []for c in channels:trace += record.annotations["abif_raw"][c][2500:15000]plt.boxplot(trace) # Aplt.axis([0,2,0,600])plt.title(record.annotations['abif_raw']['TUBE1'])plt.show()
import osimport matplotlib as mplimport matplotlib.pyplot as pltfrom Bio import SeqIOINPUT = "96Well_Result"Cmd = "ls "+ str(INPUT)LIST = os.popen(Cmd).read().split("\n")[:-1]trace = []for i in range(96):abi = INPUT+"/"+LIST[i]record = SeqIO.read(abi, "abi")channels = ["DATA1", "DATA2", "DATA3", "DATA4"]for c in channels:trace += record.annotations["abif_raw"][c][2500:15000]trace2 = []for i in range(18):abi = INPUT+"/"+LIST[i]record = SeqIO.read(abi, "abi")channels = ["DATA1", "DATA2", "DATA3", "DATA4"]for c in channels:trace2 += record.annotations["abif_raw"][c][2500:15000]trace3 = []for i in range(18,96):abi = INPUT+"/"+LIST[i]record = SeqIO.read(abi, "abi")channels = ["DATA1", "DATA2", "DATA3", "DATA4"]for c in channels:trace3 += record.annotations["abif_raw"][c][2500:15000]plt.boxplot([trace, trace2, trace3])plt.axis([0,4,0,600])
Enjoy~
由於語法渲染問題而影響閱讀體驗, 請移步博客閱讀~
本文GitPage地址
GitHub: Karobben
Blog:Karobben
BiliBili:史上最不正經的生物狗

若有收获,就点个赞吧
0 人点赞
