我们得到了 bam 文件之后,也要进行质控,可以对测序数据有更深入的理解。因此我们用几种方法来检查 bam 文件结果。
samtools stats
## stats.shcat config | while read iddobam=./4.align/${id}.bamsamtools stats -@ 16 --reference ~/wes_cancer/data/Homo_sapiens_assembly38.fasta ${bam} > ./4.align/stats/${id}.statplot-bamstats -p ./4.align/stats/${id} ./4.align/stats/${id}.statdone
这一步质控后也会生成 html 网页报告,下载到本地查看,可以看到碱基分布信息和统计信息:
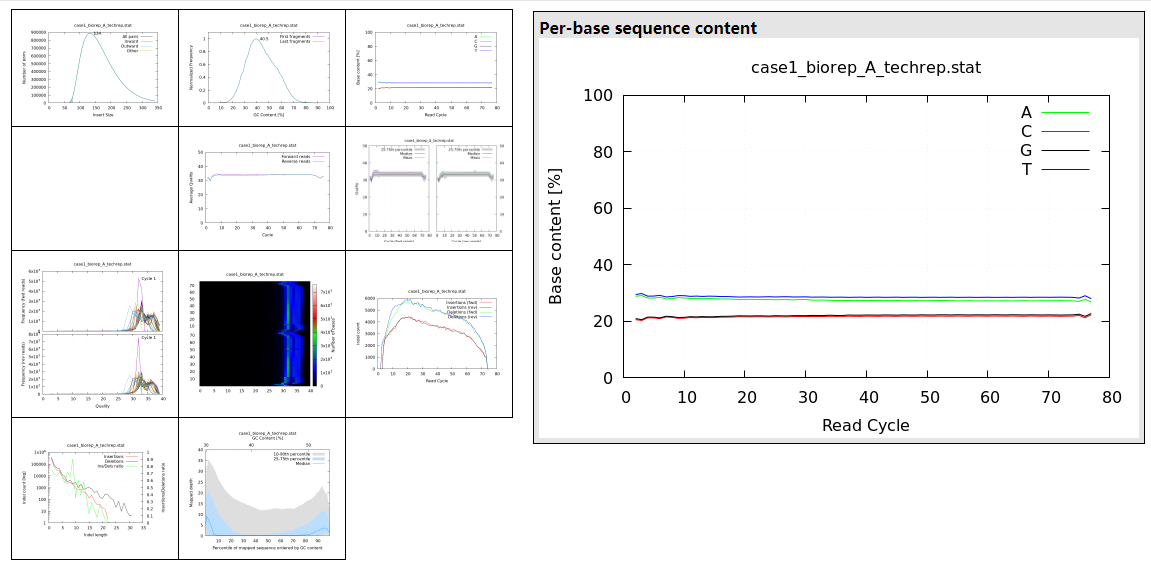
Readstotal: 212,740,888filtered: 0 (0.0%)non-primary: 21,011duplicated: 0 (0.0%)mapped: 212,738,868 (100.0%)zero MQ: 17,615,610 (8.3%)avg read length: 74Basestotal: 15,940,884,573mapped: 15,935,687,200 (100.0%)error rate: 0.20%
qualimap
接下来用 qualimap 软件来查看基因组覆盖深度等信息,代码:
cat config | while read iddoqualimap bamqc --java-mem-size=10G -gff ~/wes_cancer/data/hg38.exon.bed -nr 100000 -nw 500 -nt 16 -bam ./4.align/${id}.bam -outdir ./4.align/qualimap/${id}done
对case1_biorep_A_techrep的比对结果拿到bam文件进行统计,可以看到reads覆盖度条形图,是这样的:
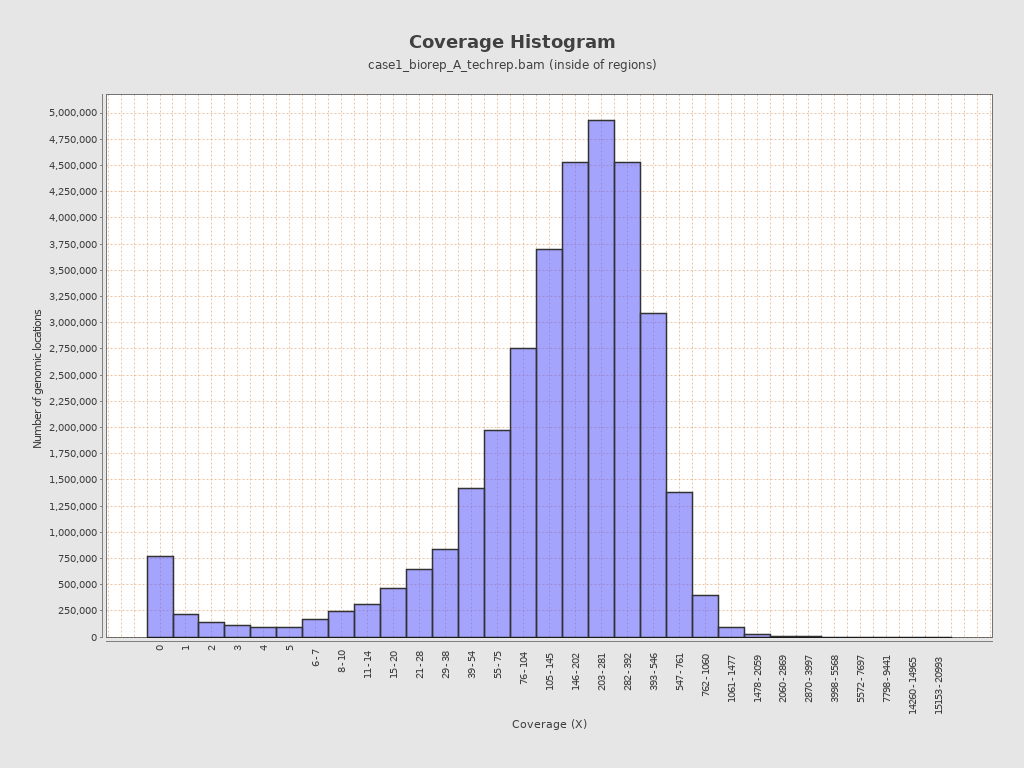
虽然只是一个样本,但从平均水平上来看,和作者在文献中说的基本符合,在文章中关于测序深度的描述是这样的:

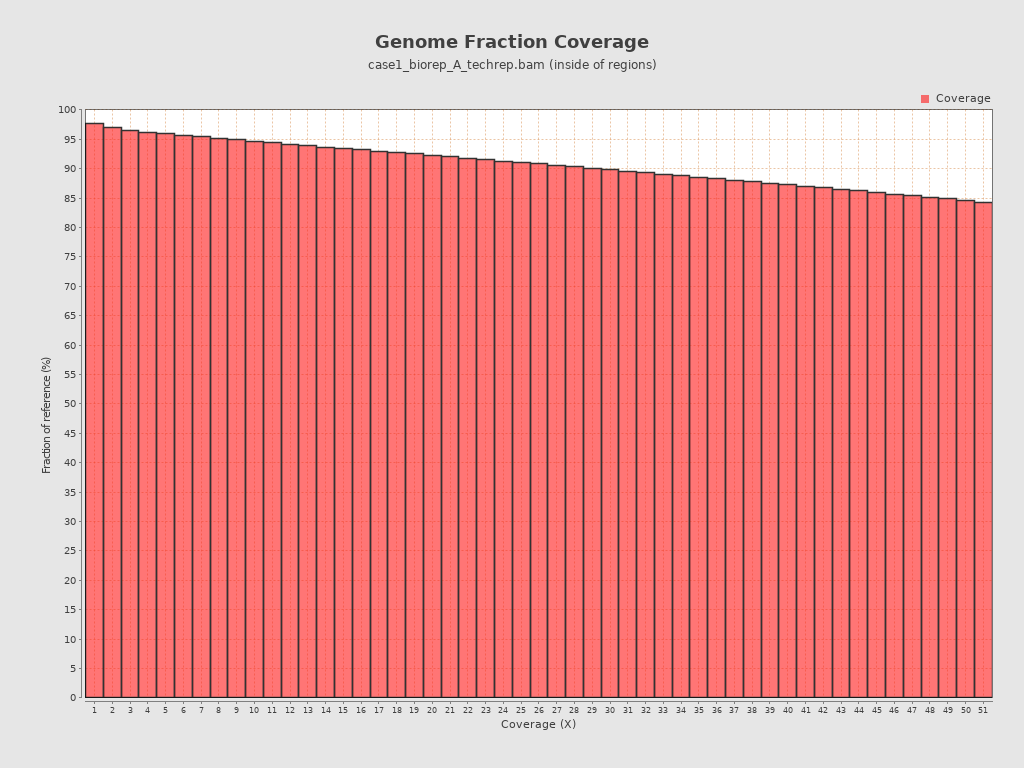
还有基因组的覆盖信息,可以看到超过85%的位点的测序深度大于50x,不过需要明确一点,这里只是外显子区域的覆盖信息,因为测序策略就是WES,检查比对结果的时候也指定了外显子坐标文件~/wes_cancer/data/hg38.exon.bed
然后就是测序前建库的 insert 插入片段的长度:
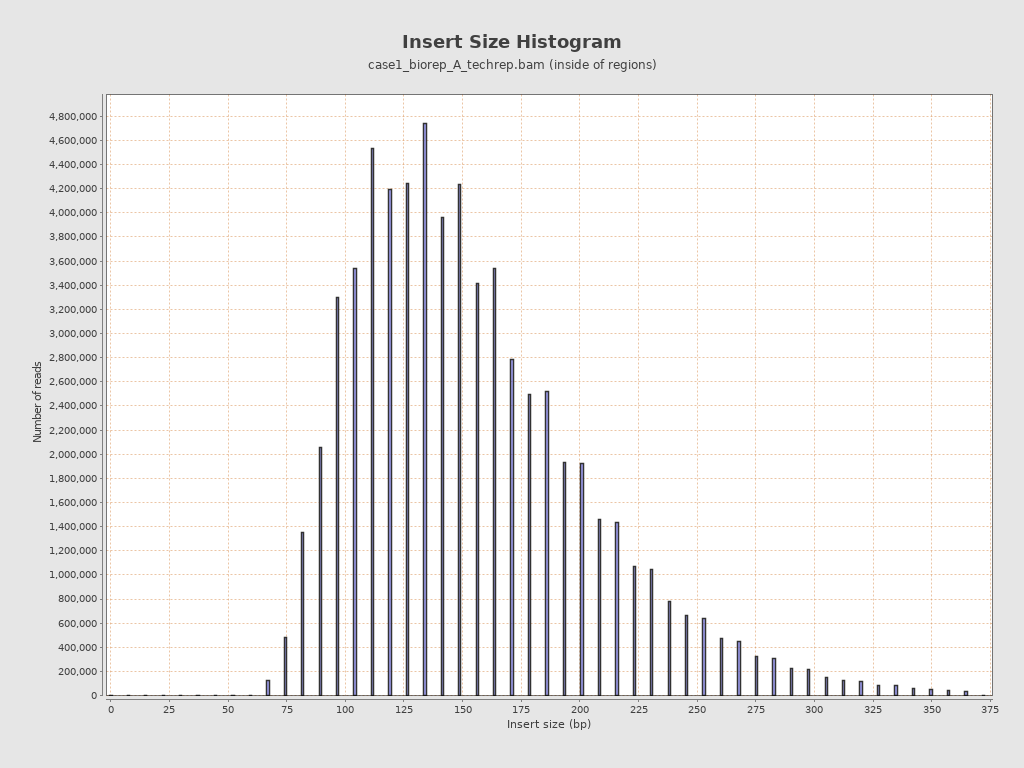
也和文献中描述的差不多:


若有收获,就点个赞吧
0 人点赞
